Hacia la electrónica molecular
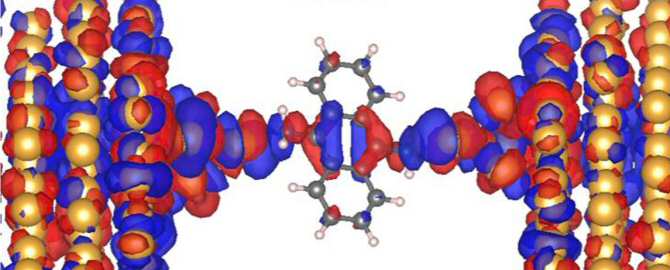
Con el desarrollo de técnicas experimentales y teóricas avanzadas, la electrónica molecular —disciplina en la que los conductores eléctricos están exclusivamente formados por moléculas orgánicas (no por metales)— ha dejado de ser una curiosidad científica para adentrarse en el mundo de los nanodispositivos funcionales.
Investigadores de la Universidad Autónoma de Madrid (UAM) y el Instituto IMDEA Nanociencia, en España ambas entidades, han demostrado que la variación de la conductividad a través de moléculas conectadas a electrodos externos, formando lo que se denomina una unión molecular, se puede predecir de manera cualitativamente correcta utilizando un modelo computacional rápido y sencillo. Esto abre las puertas el estudio masivo y sistemático de diferentes uniones moleculares, cada vez más complejas, lo que facilitará el diseño de uniones moleculares con la conductividad adecuada para cada dispositivo.
Electrónica molecular
El uso de moléculas como bloques activos en componentes electrónicos no solo puede ayudar a sobrepasar los límites establecidos por la ley de Moore en la progresiva miniaturización de dispositivos electrónicos. También ofrece nuevas funcionalidades y ventajas con respecto a la electrónica convencional.
Aun así, la funcionalidad final en este tipo de dispositivos no está definida únicamente por la naturaleza intrínseca de la molécula, sino también por la de los electrodos. Y lo más importante, por los fenómenos interfaciales entre molécula y electrodo.
Las medidas experimentales de conductancia en uniones moleculares poseen naturaleza estocástica, requiriendo un gran número de registros ante la dificultad de controlar la geometría de la unión molécula-electrodo durante las medidas. Esto da lugar a una cierta dispersión en las mediciones y requiere con frecuencia el apoyo de simulaciones teóricas para entender qué ocurre a escala nanométrica.
Modelos y supercomputación
Los métodos teóricos más utilizados para investigar este tipo de problemas son los basados en funciones de Green (FG) y en la teoría del funcional de la densidad (DFT). Estas teorías requieren de un alto nivel de conocimiento, tanto matemático como físico, siendo computacionalmente muy costosas o incluso inaccesibles con los superordenadores actuales.
En consecuencia, el avance en este campo se vería impulsado por la existencia de metodologías teóricas sencillas, rápidas y de baja demanda computacional, que además permitieran el estudio de una gran cantidad de moléculas de forma conjunta. Además, idealmente, estos modelos deberían estar al alcance de una cantidad amplia de investigadores, no solo especialistas en simulaciones computacionales, y deberían ser de fácil utilización, incluso en ordenadores personales.
El modelo propuesto por el equipo de Joel G. Fallaque se basa en la teoría de orbitales moleculares de Hückel extendida. Para comprobar su efectividad e idoneidad se aplicó sobre un total de 31 moléculas y los resultados se compararon con los de costosos cálculos DFT-S y experimentos llevados a cabo mediante la técnica de ruptura de unión usando un microscopio de efecto túnel (BJ-STM). Las simulaciones teóricas DFT-S, utilizadas para validar los resultados del modelo, se realizaron en la supercomputadora Mare Nostrum, del Centro de Supercomputación de Barcelona, requiriendo alrededor de 320 núcleos de procesador durante 30 horas.
En comparación, las simulaciones con el modelo propuesto solo necesitaron 1 núcleo y menos de 1 minuto.
Fallaque y sus colegas exponen los detalles técnicos de su modelo en la revista académica Nanoscale, bajo el título “A simple model to engineer single-molecule conductance of acenes by chemical disubstitution”. (Fuente: UAM)